Overview
Approximately 46 unrelated families worldwide have been identified with RVCL-S. Our families are located not only in the USA but also in Australia, Germany, France, the Netherlands, Spain, Mexico, Japan, China, Turkey, Switzerland and Italy.
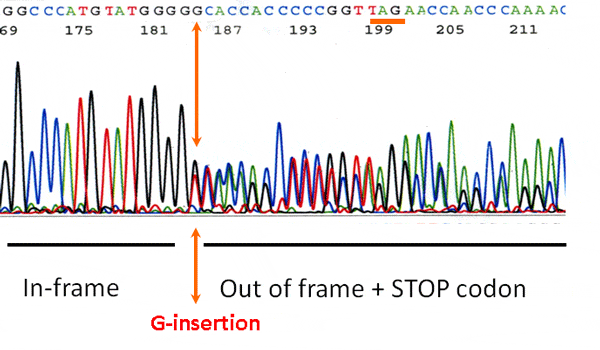
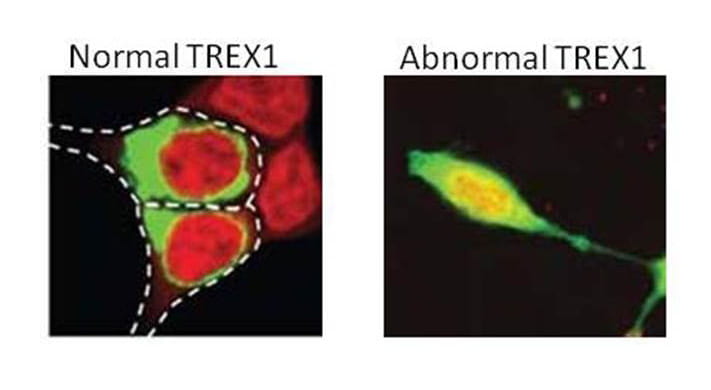
This autosomal dominant disorder features three types of mutations all of which are located at or beyond valine-235 of the 314 amino acid protein. The most dominant mutations are frame-shift mutations of TREX1 that produce a truncated protein. A second type of mutation is a Stop mutation in which the translation of TREX1 abruptly ends. Another type of mutation simply deletes amino acids in this region but it retains the amino acids after the deletion. While all of these mutation have not yet been modeled, we believe the common effect is the production of a protein that is mislocalized throughout cells instead of being anchored on the endoplasmic reticulum. We have called this aberrant localization process TREX on the loose.
Structure
The wild-type protein translated from the TREX1 gene consists of 314 amino acids (~ 32 kDa) and is encoded by a single exon on chromosome 3p21. TREX1 consists of three domains important for its role as a DNA repair enzyme (i.e., exonuclease domains I, II, and III). TREX1 also has an extended C-terminal tail domain of ~ 80 amino acids containing a leucine-rich sequence required for its endoplasmic reticulum (ER) localization.

As described above, RVCL-S results from a mutation in the tail domain that changes the protein reading frame, introduces a stop codon, and thus causes a premature termination leading to partial loss of the tail. As a result, TREX1 loses it ability to remain in the ER and becomes mislocalized throughout the cell. At present, 23 different mutations in TREX1 have been identified (Liszewski, K. & Atkinson, J.P summary as of 11/15/2022). However, of the 46 unrelated families worldwide with RVCL-S, 18 unrelated kindreds have a particular mutation identified on the protein level described as V235G fs*6. This means that in TREX1, the amino acid valine changes to a glycine at amino acid #235. Although normal TREX1 protein is 314 amino acids long, this mutation causes the protein to interrupt its normal sequence, add 6 foreign amino acids and then terminate abruptly knocking out the normal tail sequence.
For reasons that are as yet unclear, most people who carry these heterozygous TREX1 tail mutations live a fairly normal life until middle age, when vision and brain damage begin. Progressive deterioration proceeds over a 5 to 15 year period. New studies conducted by our laboratory have identified that TREX1 is expressed by a subset of human brain microglia cells that also are often close to blood vessels. However, TREX1 clusters densely in microglia cells in proximity to brain lesions of RVCL patients. Further studies are needed to determine how clustered TREX1 may impact the symptoms of RVCL.
In addition to RVCL-S, mutations in other areas of TREX1 play major roles in a variety of neurovascular and autoimmune-related disorders such as Aicardi-Goutieres syndrome, chilblain lupus, and Cree encephalitis. TREX1 mutations also have been linked to systemic lupus erythematosus (SLE) and Sjögren’s syndrome.
Related publications

Novel de novo TREX1 mutation in a patient with retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations mimicking demyelinating disease
July 1, 2021
Macaron G, Khoury J, Hajj-Ali RA, Prayson RA, Srivastava S, Ehlers JP, Mamsa H, Liszewski MK, Jen JC, Bermel RA, Ontaneda D. Novel de novo TREX1 mutation in a patient with retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations mimicking demyelinating disease. Mult Scler Relat Disord. 2021 Jul;52:103015. doi: 10.1016/j.msard.2021.103015. Epub 2021 May 7. PMID: 34044261.

Lesion evolution and neurodegeneration in RVCL-S: A monogenic microvasculopathy
October 6, 2020
Ford AL, Chin VW, Fellah S, Binkley MM, Bodin AM, Balasetti V, Taiwo Y, Kang P, Lin D, Jen JC, Grand MG, Bogacki M, Liszewski MK, Hourcade D, Chen Y, Hassenstab J, Lee JM, An H, Miner JJ, Atkinson JP. Lesion evolution and neurodegeneration in RVCL-S: A monogenic microvasculopathy. Neurology. 2020 Oct 6;95(14):e1918-e1931. doi: 10.1212/WNL.0000000000010659. Epub 2020 Sep 4. Erratum in: Neurology. 2021 May 11;96(19):919. PMID: 32887784; PMCID: PMC7682842.

Neuroimaging and cognitive profile in RVCL-S: Possible new insights into pathogenesis?
October 6, 2020
Commentary on: Lesion evolution and neurodegeneration in RVCL-S: A monogenic microvasculopathy
Dhamija R, Broomall E. Neuroimaging and cognitive profile in RVCL-S: Possible new insights into pathogenesis? Neurology. 2020 Oct 6;95(14):611-612. doi: 10.1212/WNL.0000000000010751. Epub 2020 Sep 4. PMID: 32887771.

TREX1 is expressed by microglia in normal human brain and increases in regions affected by ischemia
November 1, 2018
Kothari PH, Kolar GR, Jen JC, Hajj-Ali R, Bertram P, Schmidt RE, Atkinson JP. TREX1 is expressed by microglia in normal human brain and increases in regions affected by ischemia. Brain Pathol. 2018 Nov;28(6):806-821. doi: 10.1111/bpa.12626. Epub 2018 Oct 10. PMID: 30062819; PMCID: PMC6404532.

Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations
November 1, 2016
Stam AH, Kothari PH, Shaikh A, Gschwendter A, Jen JC, Hodgkinson S, Hardy TA, Hayes M, Kempster PA, Kotschet KE, Bajema IM, van Duinen SG, Maat-Schieman MLC, de Jong PTVM, de Smet MD, de Wolff-Rouendaal D, Dijkman G, Pelzer N, Kolar GR, Schmidt RE, Lacey J, Joseph D, Fintak DR, Grand MG, Brunt EM, Liapis H, Hajj-Ali RA, Kruit MC, van Buchem MA, Dichgans M, Frants RR, van den Maagdenberg AMJM, Haan J, Baloh RW, Atkinson JP, Terwindt GM, Ferrari MD. Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations. Brain. 2016 Nov 1;139(11):2909-2922. doi: 10.1093/brain/aww217. PMID: 27604306; PMCID: PMC5091044.

Human disease phenotypes associated with mutations in TREX1
April 1, 2015
Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. 2015 Apr;35(3):235-43. doi: 10.1007/s10875-015-0147-3. Epub 2015 Mar 4. PMID: 25731743.

Neuropathology and genetics of cerebroretinal vasculopathies
September 1, 2014
Kolar GR, Kothari PH, Khanlou N, Jen JC, Schmidt RE, Vinters HV. Neuropathology and genetics of cerebroretinal vasculopathies. Brain Pathol. 2014 Sep;24(5):510-8. doi: 10.1111/bpa.12178. PMID: 25323666; PMCID: PMC8029267.

New roles for the major human 3′-5′ exonuclease TREX1 in human disease
June 15, 2008
Kavanagh D, Spitzer D, Kothari PH, Shaikh A, Liszewski MK, Richards A, Atkinson JP. New roles for the major human 3′-5′ exonuclease TREX1 in human disease. Cell Cycle. 2008 Jun 15;7(12):1718-25. doi: 10.4161/cc.7.12.6162. Epub 2008 Jun 16. PMID: 18583934; PMCID: PMC2825026.
Function
There are two known functions for TREX1:
DNA sensor
TREX1 was originally discovered and identified as the “3 prime repair exonuclease 1” that cleans up and digests DNA debris that is generated when tissue is damaged or cells die. It degrades single-stranded DNA ~4-fold more efficiently than double-stranded DNA. TREX1 is also important for handling DNA from viruses that invade and infect cells.
Sugar sensor
A newly discovered function of TREX1 is in overseeing the ‘sugar polishing’ step (i.e., glycosylation) for newly made proteins. Thus, TREX1 interacts with cellular machinery (the oligosaccharyltransferase or “OST” complex) to add N-glycans to proteins as they are produced. These glycans are important for modulating the function and stability of proteins.
When the sugar polishing function is disrupted, TREX1 no longer can bind to its partner proteins in the OST complex. This leads to the buildup of glycans. The disruption of this important interaction also may diminish blood vessel life-span and integrity and lead to disturbances in the immune system. These studies also identified an anthracycline antibiotic (Aclarubicin) that corrected the defect in both mouse disease models and in human patient cells. Studies are underway to better understand and expand on these exciting discoveries. Additionally, human clinical trials are in development.
Related publications

Cytosolic Nuclease TREX1 Regulates Oligosaccharyltransferase Activity Independent of Nuclease Activity to Suppress Immune Activation
September 15, 2015
Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, Li QZ, Atkinson JP, Morse HC 3rd, Lehrman MA, Yan N. Cytosolic Nuclease TREX1 Regulates Oligosaccharyltransferase Activity Independent of Nuclease Activity to Suppress Immune Activation. Immunity. 2015 Sep 15;43(3):463-74. doi: 10.1016/j.immuni.2015.07.022. Epub 2015 Aug 25. PMID: 26320659; PMCID: PMC4575271.

Exonuclease TREX1 also Has a Sweet Tooth
September 15, 2015
Barchet W, Zillinger T. Exonuclease TREX1 also Has a Sweet Tooth. Immunity. 2015 Sep 15;43(3):411-3. doi: 10.1016/j.immuni.2015.08.026. PMID: 26377892.

Biochemical properties of mammalian TREX1 and its association with DNA replication and inherited inflammatory disease
June 1, 2009
Lindahl T, Barnes DE, Yang YG, Robins P. Biochemical properties of mammalian TREX1 and its association with DNA replication and inherited inflammatory disease. Biochem Soc Trans. 2009 Jun;37(Pt 3):535-8. doi: 10.1042/BST0370535. PMID: 19442247.
Disease models
Our research utilizes two types of models: human lymphoblast cell lines and mouse disease models. For the former, we prepare immortalized EBV-transformed patient B cells. Secondly, we and others have developed TREX1 mouse models that knock out of mouse TREX1 (TREX1-/-) or knock in human RVCL-S TREX1 mutation (V235fs-KI).
Parul Kothari, MD, PhD, who studied this disease at WashU Medicine, wrote her doctoral dissertation on these studies.

Studies on the Mislocalized Exonuclease TREX1 that
Causes a Systemic Vasculopathy
May 1, 2014
Kothari, Parul Harish. Studies on the Mislocalized Exonuclease TREX1 that
Causes a Systemic Vasculopathy. All Theses and Dissertations (ETDs). 1243.
https://openscholarship.wustl.edu/etd/1243.
TREX1 reagents
Our teams have produced recombinant human TREX1 and recombinant RVCL-S TREX1 as well as antibodies to normal TREX1. These reagents are valuable tools to study the disease in vitro (in the test tube) as well as in vivo (in living systems).
Researchers at the RVCL-S Research Center continue to seek a better understanding of the disease process and to find an effective treatment to replace, bypass, correct or negate the effects of defective TREX1 protein.
For additional information on RVCL-S, see our review for the National Organization for Rare Disorders (NORD).
Clinical studies and RVCL-S as a model of cerebral small vessel disease
Related publications

Lesion evolution and neurodegeneration in RVCL-S: A monogenic microvasculopathy
October 1, 2020
Ford AL, Chin VW, Fellah S, Binkley MM, Bodin AM, Balasetti V, Taiwo Y, Kang P, Lin D, Jen JC, Grand MG, Bogacki M, Liszewski MK, Hourcade D, Chen Y, Hassenstab J, Lee JM, An H, Miner JJ, Atkinson JP. Lesion evolution and neurodegeneration in RVCL-S: A monogenic microvasculopathy. Neurology. 2020 Oct 6;95(14):e1918-e1931. doi: 10.1212/WNL.0000000000010659. Epub 2020 Sep 4. Erratum in: Neurology. 2021 May 11;96(19):919. PMID: 32887784; PMCID: PMC7682842.